

Cardiology
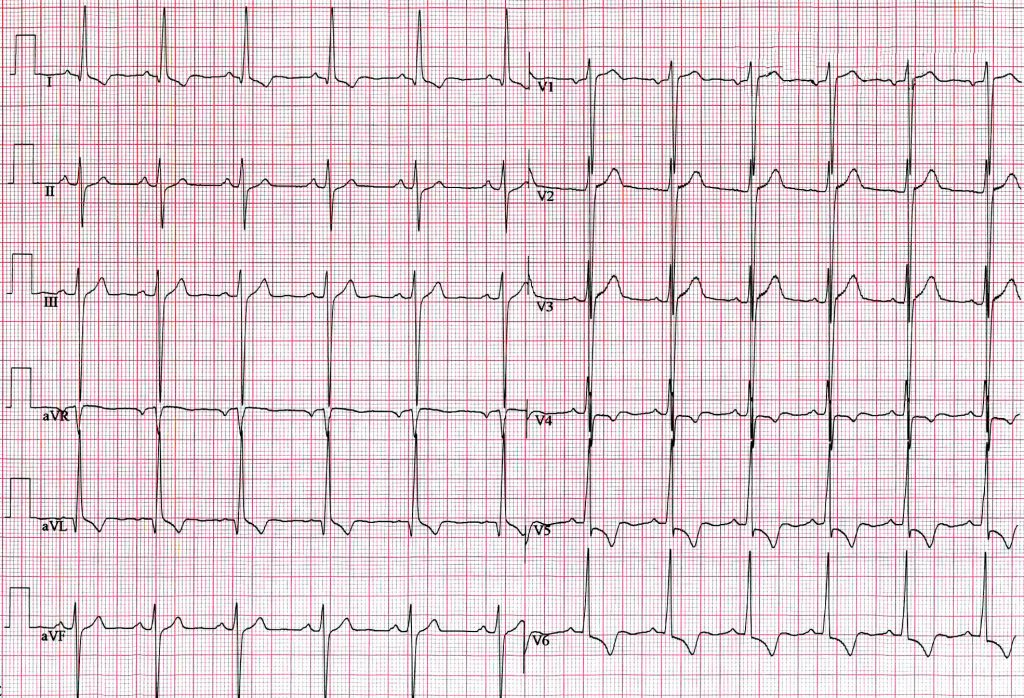
A 19 year old man presents to the Emergency Department after collapsing whilst running. He tells you this is the second time he has "blacked out" whilst running - he did not present to medical services on the first occasion. He also reports several episodes of palpitations and dizzy spells over the previous 3 months. He has no significant past medical history. His father died suddenly aged 30 years. On examination you note a harsh ejection systolic murmur at the left sternal edge. His observations are recorded as:
- Heart rate: 80 beats/minute
- Blood pressure: 132/76 mmHg
- Respiratory rate: 18 breaths/minute
- Oxygen saturations: 98% on air
His ECG is shown. What is the most likely diagnosis?
Answer:
The ECG shows tall QRS complexes with ST depression and deep T-wave inversions in the lateral leads. These features are quite classical of the presence of left ventricular hypertrophy (LVH). With a family history of sudden death, the boy’s recurrent episodes of syncope and auscultation of ejection systolic murmur point firmly towards the diagnosis of hypertrophic cardiomyopathy (HCM). It is the most common genetic heart disease as well as the most frequent cause of sudden cardiac death in young people.Cardiomyopathies
Cardiology
Last Updated: 8th September 2021
The cardiomyopathies are an important, heterogeneous group of heart muscle diseases that make a significant contribution to morbidity and mortality. They are associated with mechanical and/or electrical dysfunction. Inappropriate ventricular hypertrophy or dilatation is usually present. Cardiomyopathy involvement may be predominantly limited to the heart (primary cardiomyopathy) or form part of a generalised systemic disorder (secondary cardiomyopathy). Causes vary widely, but genetic aetiologies are most common in primary cardiomyopathies. Complications include cardiovascular death and progressive heart failure, with its associated disability.
Causes
Primary cardiomyopathies:
- Genetic
- Hypertrophic cardiomyopathy (HCM)
- Hypertrophic cardiomyopathy (HCM) is a genetic disorder characterised by the development of a hypertrophied, non-dilated left ventricle (LV) in the absence of another predisposing condition (such as aortic stenosis or hypertension). It is the most common genetic heart disease as well as the most frequent cause of sudden cardiac death in young people.
- Up to one-third of patients have resting left ventricular outflow obstruction, while others may have exercise or other haemodynamic strain-induced outflow gradients. Up to 10% of individuals will develop end-stage systolic dysfunction, often in the absence of significant dilatation. Atrial arrhythmias are common, and affected individuals have a high risk of thromboembolism.
- Many patients will have no symptoms at the time of diagnosis and will be diagnosed following routine examination or family screening of an affected family member. The diagnosis is usually made using a combination of ECG and cardiac imaging, most commonly echocardiography.
- Medical therapy with beta-blockers, calcium-channel blockers, or disopyramide is used in symptomatic patients. A subset of patients with increased risk for sudden death should undergo defibrillator implantation.
- Arrhythmogenic right ventricular cardiomyopathy (ARVC)
- Characterised by progressive replacement of myocardium with fibro-fatty material. These changes are found most commonly in the triangle of dysplasia (right ventricular inflow, outflow, and apex) but are increasingly recognised in the LV.
- ARVC represents a wide spectrum of disease, involving both electrophysiological and functional abnormalities. Electrophysiological manifestations range from the most modest of ECG abnormalities to life-threatening ventricular arrhythmias. Similarly, abnormalities of muscle function range from minor contractile changes to severe right and left ventricular failure. Patients present with a wide variety of symptoms ranging from arrhythmias to heart failure to sudden death.
- Diagnosis of ARVC is difficult and requires use of multiple investigations, including: imaging with echocardiography/MRI/angiography, ambulatory monitoring and exercise stress to look for arrhythmias, presence of abnormal resting ECG (T wave abnormalities, delayed conduction in the right-sided leads, epsilon waves), late potential on signal-averaged ECG, and histopathological changes seen on endomyocardial biopsy or at postmortem.
- Left ventricular non-compaction
- This condition is characterised by the presence of spongy myocardium, thought to be due to an arrest in normal embryogenesis (compaction starts from weeks 5 to 8 and is more pronounced in the left than right ventricle). Left ventricular non-compaction predominantly involves the apical portion of the LV chamber.
- The diagnosis is usually established on cardiac imaging using echocardiography, cardiac MRI, or LV angiography. In some patients LV dilatation and systolic dysfunction may develop. Patients may also develop dysrhythmias including sudden death, and they have an increased risk of thromboembolism, in part relating to thrombi development in the areas of non-compaction.
- Hypertrophic cardiomyopathy (HCM)
- Mixed
- Dilated cardiomyopathy (DCM)
- Characterised by the presence of left ventricular dilatation and systolic dysfunction in the absence of abnormal loading conditions or significant coronary artery disease. Right ventricular dilatation is often also present. It is estimated that 25% to 35% of cases are familial.
- An extensive list of conditions that may lead to dilated cardiomyopathy has been recognised. One well-known cause is post-myocarditis, which is often asymptomatic from a patient's perspective. Other causes include: alcohol; chemotherapeutic agents; storage diseases; autoimmune and systemic disorders; neuromuscular disorders; mitochondrial, metabolic/endocrine, and nutritional disorders. If no cause is identified, despite extensive investigation, the term idiopathic dilated cardiomyopathy is applied.
- Restrictive cardiomyopathy (RCM)
- A less well-defined cardiomyopathy as its diagnosis is based on establishing the presence of a restrictive ventricular filling pattern. The European Society of Cardiology Working Group defines it as: 'restrictive physiology in the presence of normal or reduced diastolic volumes, normal or reduced systolic volumes and normal wall thickness'. It may be idiopathic, familial, or associated with various systemic disorders.
- Patients may present with symptoms of dyspnoea or palpitations, and these relate to diastolic dysfunction. There is usually an elevated jugular venous pressure. The ECG is typically abnormal with evidence of bi-atrial hypertrophy and non-specific ST-T wave changes. Cardiac imaging by echocardiography or MRI shows bi-atrial enlargement with evidence of diastolic dysfunction, evident on mitral inflow Doppler patterns as well as tissue Doppler signals.
- As it may present in a similar manner to constrictive pericarditis, restrictive cardiomyopathy must be distinguished from that condition, and this may require cardiac catheterisation. Endomyocardial biopsy may be helpful in selected cases.
- Dilated cardiomyopathy (DCM)
- Acquired
- Inflammatory myocarditis
- Stress provoked (tako-tsubo)
- Peripartum
- Tachycardia-induced
Secondary cardiomyopathies:
- Infiltrative or storage diseases e.g. amyloidosis, Gaucher's disease, mucopolysaccharidoses, haemochromatosis, Fabry's disease, Pompe's disease, Niemann-Pick's disease
- Toxins e.g. doxorubicin, trastuzumab, chronic alcohol consumption, cobolt, heavy metals
- Endocrine disorders e.g. diabetes mellitus, hyperthyroidism/hypothyroidism, hyperparathyroidism, phaeochromocytoma, acromegaly
- Nutritional deficiencies e.g. thiamine deficiency, other nutritional deficiencies involving vitamin C, niacin, selenium, and vitamin D
- Neuromuscular and neurological disorders e.g. Friedreich's ataxia, muscular dystrophies (Duchenne, Becker, and Emery-Dreifuss muscular dystrophy; myotonic dystrophy)
- Autoimmune disorders e.g. systemic lupus erythematosus
- Sarcoidosis
- Electrolyte imbalance e.g. potassium, phosphate, or magnesium disturbance
- Endomyocardial fibrosis
- Loeffler's endocarditis/hypereosinophilic syndrome
Clinical features
- History
- The clinical history is a critical component in the diagnosis of cardiomyopathies. Emphasis should be placed on risk factors for specific causes. Medical illnesses, family history, and alcohol and drug exposure may predispose to the development of cardiomyopathies.
- The most common symptoms associated with cardiomyopathy are dyspnoea, chest discomfort, palpitations, and syncope.
- Patients with concerning cardiac symptoms such as exertional chest pain, syncope, sustained palpitations, orthopnoea, paroxysmal nocturnal dyspnoea, and those with a family history of cardiomyopathy, arrhythmia or premature cardiac death should be referred for specialist investigation.
- Examination
- No specific physical examination findings are consistent with a particular cause; the examination is directed towards seeking signs of cardiac dysfunction.
- A sustained prominent apical impulse on palpation may suggest left ventricular hypertrophy. A diffusely palpable cardiac impulse with apical displacement may be seen with ventricular dilatation.
- Auscultation of the heart may reveal murmurs, an S4 gallop (heard in types of cardiomyopathy that involve increased left ventricular pressure) or an S3 gallop (heard when there is increased left ventricular volume).
- Auscultation of the lungs may demonstrate crackles, indicating pulmonary congestion.
- Pedal and leg oedema, and jugular venous distension, may also be present in certain cardiomyopathies.
- Specialist referral is indicated if any of the following features are present: an ejection systolic murmur (in left ventricular outflow tract obstruction, as seen in hypertrophic cardiomyopathy, this increases in intensity with the Valsalva manoeuvre and is diminished by squatting); peripheral and/or pulmonary oedema; elevated jugular venous pressure; an irregular pulse.
Investigations
- ECG
- An ECG, although commonly non-specific, may show abnormalities that point to an individual aetiology. A normal ECG has, approximately, a 98% negative predictive value when systolic dysfunction is suspected.
- Laboratory tests
- Initial laboratory tests should include baseline blood tests such as FBC, metabolic panel, and thyroid function in all patients to exclude contributory or exacerbating factors. A B-type natriuretic peptide provides supporting data in patients with dyspnoea. Patients presenting with chest pain require measurement of cardiac markers including troponins and creatine kinase myocardial band (CK-MB). In addition, a chest x-ray is recommended.
- Echocardiogram
- Echocardiography is an extremely useful and non-invasive diagnostic tool. An echocardiogram helps to distinguish the cardiomyopathies with respect to pathophysiology. It can differentiate between hypertrophic, restrictive, or dilated cardiomyopathies in most cases. It may suggest a diagnosis of arrhythmogenic right ventricular (ARVC) or athlete's heart.
- Further imaging
- Newer available imaging modalities include cardiac magnetic resonance imaging (MRI) and cardiac CT. Cardiac MRI is a useful method for uncovering the aetiology of cardiomyopathy because it provides excellent contrast and spatial resolution of the heart. MRI can be used to assess ventricular end-diastolic volumes, presence of intracardiac thrombi, stroke volume, ejection fraction, and valvular pathology. Cardiac CT is particularly helpful in the assessment of possible concomitant coronary artery disease.
- Cardiac catheterisation may be performed if the results of echocardiography are ambiguous. Ventricular and atrial pressures are measured, which allow for the calculation of pressure gradients across cardiac valves and the left ventricular outflow tract. Catheterisation of the heart allows for ventriculography to be performed. Ejection fraction, ventricular size and wall motion, and left ventricular outflow tract size can be estimated, and the presence of valvular regurgitation can be evaluated. Additionally, coronary angiography can be performed to evaluate for coronary arterial disease as a cause of ischaemia.
- Endomyocardial biopsy (EBM)
- In very rare circumstances, an endomyocardial biopsy is needed to help differentiate disease processes and guide therapy. It may be useful in the diagnosis of certain types of cardiomyopathy (e.g. infiltrative), in particular where there has been a recent onset of severe heart failure.
- Other specialised testing
- Testing may be directed towards excluding possible causes of cardiomyopathy
Causes of sudden cardiac death (SCD)
Sudden cardiac death (SCD) is an unexpected death due to cardiac causes that occurs in a short time period (generally within 1 hour of symptom onset) in a person with known or unknown cardiac disease. Most cases of SCD are related to cardiac arrhythmias. Risk factors include:
- Ischaemic heart disease
- Myocardial disease
- Hypertrophic cardiomyopathy (HCM)
- Dilated cardiomyopathy (DCM)
- Arrhythmogenic right ventricular cardiomyopathy (ARVC)
- Restrictive cardiomyopathy (RCM)
- Myocarditis
- Valvular heart disease
- Aortic stenosis
- Aortic insufficiency
- Mitral stenosis
- Mitral valve prolapse
- Congenital heart disease
- Tetralogy of Fallot
- Transposition of the great arteries
- Fontan operation
- Aortic stenosis
- Marfan syndrome
- Mitral valve prolapse
- Hypoplastic left heart syndrome
- Eisenmenger syndrome
- Congenital heart block
- Ebstein anomaly
- Ion channelopathies
- Long QT syndrome (LQTS)
- Brugada syndrome
- Progressive cardiac conduction defect (PCCD)
- Catecholaminergic polymorphic ventricular tachycardia (CPVT)
- Short QT syndrome (SQTS)
- Idiopathic ventricular fibrillation (without Brugada ECG changes)
- Other
- Wolff-Parkinson-White syndrome
Report A Problem
Is there something wrong with this question? Let us know and we’ll fix it as soon as possible.
Loading Form...
- Biochemistry
- Blood Gases
- Haematology
| Biochemistry | Normal Value |
|---|---|
| Sodium | 135 – 145 mmol/l |
| Potassium | 3.0 – 4.5 mmol/l |
| Urea | 2.5 – 7.5 mmol/l |
| Glucose | 3.5 – 5.0 mmol/l |
| Creatinine | 35 – 135 μmol/l |
| Alanine Aminotransferase (ALT) | 5 – 35 U/l |
| Gamma-glutamyl Transferase (GGT) | < 65 U/l |
| Alkaline Phosphatase (ALP) | 30 – 135 U/l |
| Aspartate Aminotransferase (AST) | < 40 U/l |
| Total Protein | 60 – 80 g/l |
| Albumin | 35 – 50 g/l |
| Globulin | 2.4 – 3.5 g/dl |
| Amylase | < 70 U/l |
| Total Bilirubin | 3 – 17 μmol/l |
| Calcium | 2.1 – 2.5 mmol/l |
| Chloride | 95 – 105 mmol/l |
| Phosphate | 0.8 – 1.4 mmol/l |
| Haematology | Normal Value |
|---|---|
| Haemoglobin | 11.5 – 16.6 g/dl |
| White Blood Cells | 4.0 – 11.0 x 109/l |
| Platelets | 150 – 450 x 109/l |
| MCV | 80 – 96 fl |
| MCHC | 32 – 36 g/dl |
| Neutrophils | 2.0 – 7.5 x 109/l |
| Lymphocytes | 1.5 – 4.0 x 109/l |
| Monocytes | 0.3 – 1.0 x 109/l |
| Eosinophils | 0.1 – 0.5 x 109/l |
| Basophils | < 0.2 x 109/l |
| Reticulocytes | < 2% |
| Haematocrit | 0.35 – 0.49 |
| Red Cell Distribution Width | 11 – 15% |
| Blood Gases | Normal Value |
|---|---|
| pH | 7.35 – 7.45 |
| pO2 | 11 – 14 kPa |
| pCO2 | 4.5 – 6.0 kPa |
| Base Excess | -2 – +2 mmol/l |
| Bicarbonate | 24 – 30 mmol/l |
| Lactate | < 2 mmol/l |