

Haematology
This was previously featured in an exam
An 8 year old boy, with known haemophilia A, is brought to ED with a scalp wound which is bleeding profusely. You are planning for blood transfusion. Which of the following blood products is the priority in this patient?
Answer:
- Single-factor concentrates are available for the treatment of most inherited coagulation deficiencies except Factor V and Factor II (prothrombin). Most patients in the UK with severe haemophilia A are now treated with recombinant Factor VIIIc, which carries no risk of viral or prion transmission.
- FFP is indicated for the treatment of patients with bleeding due to multiple clotting factor deficiencies such as disseminated intravascular coagulation (DIC). It may also be used in patients with inherited clotting factor deficiencies (e.g. Factor V deficiency) where a clotting factor concentrate is not yet available.
- Cryoprecipitate is made by thawing UK donor FFP at 4°C, producing a cryoglobulin rich in fibrinogen, Factor VIII and von Willebrand factor. It was developed as a treatment for haemophilia but this use has now been replaced by Factor VIII concentrate. Cryoprecipitate is mainly used as a more concentrated, hence lower volume for infusion, source of fibrinogen than FFP.
Haemophilia
Haematology
Last Updated: 8th September 2021
Haemophilia is a bleeding disorder, usually inherited with an X-linked recessive inheritance pattern, which results from the deficiency of a coagulation factor. Haemophilia A results from the deficiency of clotting factor VIII. Haemophilia B results from the deficiency of clotting factor IX. Acquired haemophilia is a separate non-inherited condition. It is much rarer than congenital haemophilia and has an autoimmune-related aetiology with no genetic inheritance pattern.
Inheritance pattern
Congenital haemophilia has an X-linked recessive pattern of inheritance. Therefore, boys/men are exclusively affected, although many female carriers have clotting factor levels in the haemophilia range due to lyonization (random inactivation of the normal X chromosome) and may have bleeding symptoms requiring appropriate management. Up to one third of patients with congenital haemophilia have no family history, as the condition may also result from spontaneous germline mutations and/or somatic mosaicism during early embryogenesis
In an X-linked recessive pattern (assuming the other parent is a non-carrier):
- Sons of a female carrier have a 50% chance of developing the disease
- Daughters of a female carrier have a 50% chance of being a carrier
- All sons of a male haemophiliac will be unaffected
- All daughters of a male haemophiliac will be a carrier
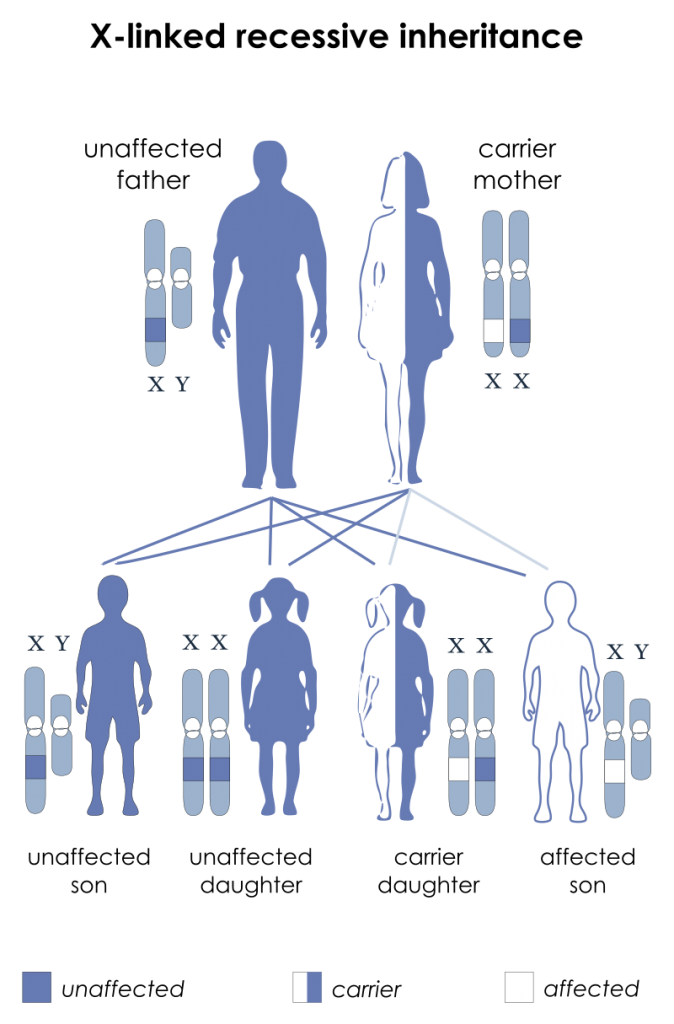
X-linked Recessive Inheritance Pattern. (Image by National Institutes of Health derivative work: Drsrisenthil [Public domain])
Clinical features
A characteristic personal and family history, along with physical examination findings, raises suspicion for the diagnosis of congenital haemophilia. This is subsequently confirmed by laboratory findings. The age of presentation and bleeding frequency are influenced by the severity of the condition. Most patients are diagnosed as children. However, some patients may go undiagnosed until adulthood, particularly those with mild or even moderate haemophilia who have had no significant haemostatic challenges in their lives. Haemophilia A and B are clinically indistinguishable.
Typical symptoms of haemophilia:
- Recurrent or severe bleeding symptoms, or bleeding in unusual sites (e.g. joints or muscles).
- Minor mucocutaneous bleeding (e.g. epistaxis, bleeding from gums following minor dental procedures, easy bruising) as well as severe bleeding following trauma, surgery, or dental procedures, are commonly described.
- Musculoskeletal bleeding is the hallmark of haemophilia. Presents with pain and swelling of the involved area, commonly extremities, with decreased range of motion, erythema, and increased local warmth.
- Gastrointestinal bleeding and haematuria are seen in patients with congenital haemophilia. They may occur spontaneously or following trauma. Women and girls who are carriers of congenital haemophilia, particularly those with clotting factor levels in the haemophilia range, most commonly present with menorrhagia and bleeding following surgical procedures or childbirth.
Typical signs of childhood or adult presentation include:
- Active bleeding from a site of injury
- Excessive bruising or haematoma formation
- Joint pain/swelling (common joints include knee, ankle and elbow)
- Extremity pain/swelling
- Decreased range of motion of an extremity
- Distended and painful abdomen
- Pallor
- Focal neurological deficits
- Haematuria
Investigations
- Full blood count
- To rule out thrombocytopenia and to identify anaemia
- Coagulation studies
- Activated partial thromboplastin time (aPTT)
- To confirm prolonged APTT
- Factor VIII and IX assay
- To confirm low levels
- Prothrombin time
- To evaluate the extrinsic and common pathways of coagulation
- Closure time/bleeding time and platelet aggregation studies
- To evaluate platelet function
- Von Willebrand factor studies
- To exclude von Willebrand disease
- Other specific factor assays:
- As needed, based on PT and aPTT results
- Activated partial thromboplastin time (aPTT)
- LFTs
- To evaluate for liver dysfunction that can also contribute to prolongation of PT and aPTT
- Imaging studies or endoscopy may be required for evaluation of acute bleeding
Management
Comprehensive care for congenital haemophilia involves:
- Treatment and prevention of bleeding
- Long-term management of joint and muscle damage, and of other sequelae of bleeding
- Management and prevention of complications from treatment
- Education and promotion of self-guided care
- Home therapy
- Physiotherapy and joint status-appropriate exercises for muscle strengthening, joint protection, maintenance of range of motion, and balance.
Management of acute bleeding episodes:
- Treatment options for most patients with congenital haemophilia consist of factor VIII or factor IX replacement (for haemophilia A and haemophilia B, respectively) by infusion of factor VIII or IX concentrate.
- Additional treatments include:
- Antifibrinolytic agents (e.g. tranexamic acid, aminocaproic acid)
- Pain medications
- Desmopressin: patients with mild haemophilia A (with a demonstrated positive response to desmopressin) may benefit.
- Topical haemostatic agents such as fibrin sealant and topical thrombin are used primarily in the surgical setting.
Prophylaxis:
- Prophylactic therapy is indicated in most patients with severe haemophilia A or B. It is defined as regular, continuous intravenous factor replacement (factor VIII for haemophilia A, factor IX for haemophilia B) given for at least 45 weeks/year in anticipation of, and to prevent, bleeding.
Report A Problem
Is there something wrong with this question? Let us know and we’ll fix it as soon as possible.
Loading Form...
- Biochemistry
- Blood Gases
- Haematology
| Biochemistry | Normal Value |
|---|---|
| Sodium | 135 – 145 mmol/l |
| Potassium | 3.0 – 4.5 mmol/l |
| Urea | 2.5 – 7.5 mmol/l |
| Glucose | 3.5 – 5.0 mmol/l |
| Creatinine | 35 – 135 μmol/l |
| Alanine Aminotransferase (ALT) | 5 – 35 U/l |
| Gamma-glutamyl Transferase (GGT) | < 65 U/l |
| Alkaline Phosphatase (ALP) | 30 – 135 U/l |
| Aspartate Aminotransferase (AST) | < 40 U/l |
| Total Protein | 60 – 80 g/l |
| Albumin | 35 – 50 g/l |
| Globulin | 2.4 – 3.5 g/dl |
| Amylase | < 70 U/l |
| Total Bilirubin | 3 – 17 μmol/l |
| Calcium | 2.1 – 2.5 mmol/l |
| Chloride | 95 – 105 mmol/l |
| Phosphate | 0.8 – 1.4 mmol/l |
| Haematology | Normal Value |
|---|---|
| Haemoglobin | 11.5 – 16.6 g/dl |
| White Blood Cells | 4.0 – 11.0 x 109/l |
| Platelets | 150 – 450 x 109/l |
| MCV | 80 – 96 fl |
| MCHC | 32 – 36 g/dl |
| Neutrophils | 2.0 – 7.5 x 109/l |
| Lymphocytes | 1.5 – 4.0 x 109/l |
| Monocytes | 0.3 – 1.0 x 109/l |
| Eosinophils | 0.1 – 0.5 x 109/l |
| Basophils | < 0.2 x 109/l |
| Reticulocytes | < 2% |
| Haematocrit | 0.35 – 0.49 |
| Red Cell Distribution Width | 11 – 15% |
| Blood Gases | Normal Value |
|---|---|
| pH | 7.35 – 7.45 |
| pO2 | 11 – 14 kPa |
| pCO2 | 4.5 – 6.0 kPa |
| Base Excess | -2 – +2 mmol/l |
| Bicarbonate | 24 – 30 mmol/l |
| Lactate | < 2 mmol/l |